近日,南京航空航天大学郭万林院士团队的张助华教授联合美国内华达大学拉斯维加斯分校陈长风教授基于第一性原理高通量计算与键合分析揭示了过渡金属硫族化合物(TMD)的自插层机制,该研究成果以“Mechanism Regulating Self-Intercalation in Layered Materials”为题在线发表在Nano Letters上。

该研究团队首先通过第一性原理团簇展开方法(CE)以2H和3R堆垛的双层TaS2为例进行了大规模结构搜索,结果表明插层的Ta原子可表现为单分散相或三聚体相,随着插层Ta原子覆盖浓度的增加,三聚体相可能进一步演变为四聚体相和六方相。对Ta-Ta键和Ta-S键的分析表明这种相多样性源于vdW间隙内Ta-Ta和Ta-TaS2两种相互作用的竞争。为了更深入地理解自插层机制,团队基于密度泛函理论(DFT)对15种自插层过渡金属硫族化合物MX2 (si-MX2, M=transition metal and X=S, Se, Te, etc.)双分子层进行了高通量计算。获得的2322组能量数据表明,在自插层二维材料体系中普遍存在类似浓度依赖的结构相,并发现插层原子与MX2双层之间的结合能是一关键描述符,可准确预测给定si-MX2的最稳定相。这些结果被基于第一性原理的分子动力学模拟证实,并与最近的大量实验结果相一致。
这项工作揭示了TMD材料家族中金属原子自插层的机制,同时为通过插层工程合理设计二维材料的结构和性能提供了准则。
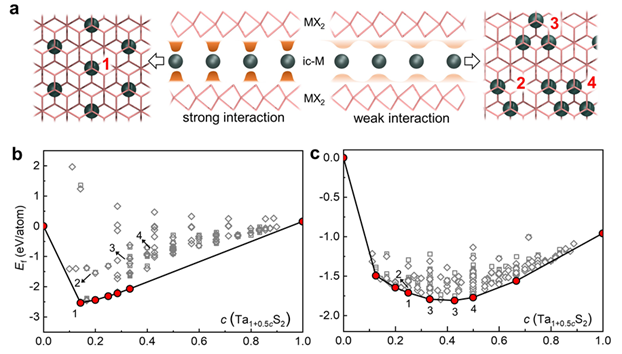
图1 si-MX2双分子层的相多样性。(a) 自插层M原子(ic-M)的示意图,左边为单体,右边为二聚体、三聚体和四聚体。(b) 3R堆垛的si-TaS2的ic-Ta原子形成能Ef随插层浓度变化的包络曲线。(c) 2H堆垛的si-TaS2的ic-Ta原子形成能Ef随插层浓度变化的包络曲线。
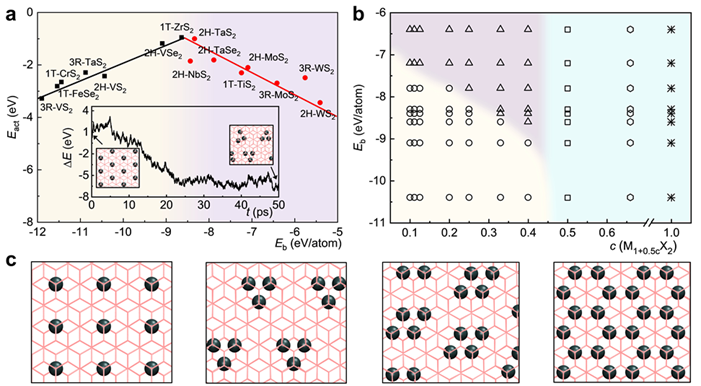
图2 si-MX2双分子层火山曲线及相图。(a) c=33%时的活化能火山曲线。插图展示了在MD模拟中si-WS2由单体向三聚体的转变。(b) si-MX2双分子层相图。(c) 单体、三聚体、四聚体和六方相的原子结构。
论文第一作者为博士生张培琨,郭万林院士、陈长风教授与张助华教授为共同通讯作者,共同作者还包括薛敏珉博士。该项工作得到了国家重点研发计划、国家自然科学基金、江苏省自然科学基金等的资助。部分计算在南京航空航天大学高性能计算中心完成。
论文链接:https://pubs.acs.org/doi/10.1021/acs.nanolett.3c00827
 学校首页
学校首页 English
English